Pharmacology: Pharmacodynamics: Mechanism of Action: Lansoprazole belongs to a class of antisecretory compounds, the substituted benzimidazoles, that suppress gastric acid secretion by specific inhibition of the (H
+, K
+)-ATPase enzyme system at the secretory surface of the gastric parietal cell. Because this enzyme system is regarded as the acid (proton) pump within the parietal cell, lansoprazole has been characterized as a gastric acid-pump inhibitor, in that it blocks the final step of acid production. This effect is dose-related and leads to inhibition of both basal and stimulated gastric acid secretion irrespective of the stimulus.
Lansoprazole does not exhibit anticholinergic or histamine type-2 antagonist activity.
Pharmacodynamic effects: Antisecretory Activity: After oral administration, lansoprazole was shown to significantly decrease the basal acid output and significantly increase the mean gastric pH and percent of time the gastric pH was greater than three and greater than four. Lansoprazole also significantly reduced meal-stimulated gastric acid output and secretion volume, as well as pentagastrin-stimulated acid output. In patients with hypersecretion of acid, lansoprazole significantly reduced basal and pentagastrin-stimulated gastric acid secretion. Lansoprazole inhibited the normal increases in secretion volume, acidity and acid output induced by insulin.
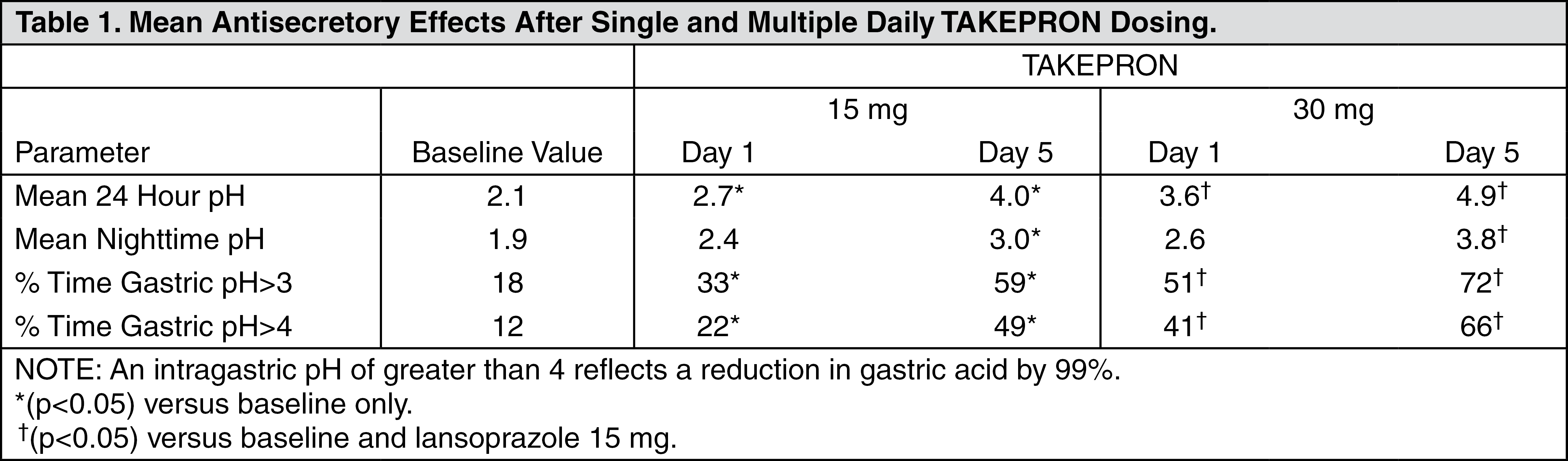
The intragastric pH results of a five-day, pharmacodynamic, crossover study of 15 mg and 30 mg of once daily lansoprazole are presented in Table 1: (See Table 1.)
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
After the initial dose in this study, increased gastric pH was seen within one to two hours with 30 mg of lansoprazole and two to three hours with 15 mg of lansoprazole. After multiple daily dosing, increased gastric pH was seen within the first hour post-dosing with 30 mg of lansoprazole and within one to two hours post-dosing with 15 mg of lansoprazole.
Acid suppression may enhance the effect of antimicrobials in eradicating
Helicobacter pylori (
H. pylori). The percentage of time gastric pH was elevated above five and six was evaluated in a crossover study of TAKEPRON given daily, twice daily and three times daily (see Table 2).
Click on icon to see table/diagram/image
The inhibition of gastric acid secretion as measured by intragastric pH gradually returned to normal over two to four days after multiple doses.There was no indication of rebound gastric acidity.
Enterochromaffin-like (ECL) Cell Effects: During lifetime exposure of rats with up to 150 mg/kg/day of lansoprazole dosed seven days per week, marked hypergastrinemia was observed followed by ECL cell proliferation and formation of carcinoid tumors, especially in female rats. Gastric biopsy specimens from the body of the stomach from approximately 150 patients treated continuously with lansoprazole for at least one year did not show evidence of ECL cell effects similar to those seen in rat studies. Longer term data are needed to rule out the possibility of an increased risk of the development of gastric tumors in patients receiving long- term therapy with lansoprazole (see Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility in the following text).
Other Gastric Effects in Humans: Lansoprazole did not significantly affect mucosal blood flow in the fundus of the stomach. Due to the normal physiologic effect caused by the inhibition of gastric acid secretion, a decrease of about 17% in blood flow in the antrum, pylorus, and duodenal bulb was seen. Lansoprazole significantly slowed the gastric emptying of digestible solids. Lansoprazole increased serum pepsinogen levels and decreased pepsin activity under basal conditions and in response to meal stimulation or insulin injection. As with other agents that elevate intragastric pH, increases in gastric pH were associated with increases in nitrate-reducing bacteria and elevation of nitrite concentration in gastric juice in patients with gastric ulcer. No significant increase in nitrosamine concentrations was observed.
Serum Gastrin Effects: In over 2100 patients, median fasting serum gastrin levels increased 50% to 100% from baseline but remained within normal range after treatment with 15 to 60 mg of oral lansoprazole. These elevations reached a plateau within two months of therapy and returned to pretreatment levels within four weeks after discontinuation of therapy.
Endocrine Effects: Human studies for up to one year have not detected any clinically significant effects on the endocrine system. Hormones studied include testosterone, luteinizing hormone (LH), follicle stimulating hormone (FSH), sex hormone binding globulin (SHBG), dehydroepiandrosterone sulfate (DHEA-S), prolactin, cortisol, estradiol, insulin, aldosterone, parathormone, glucagon, thyroid stimulating hormone (TSH), triiodothyronine (T3), thyroxine (T4), and somatotropic hormone (STH). Lansoprazole in oral doses of 15 to 60 mg for up to one year had no clinically significant effect on sexual function. In addition, lansoprazole in oral doses of 15 to 60 mg for two to eight weeks had no clinically significant effect on thyroid function. In 24-month carcinogenicity studies in Sprague-Dawley rats with daily lansoprazole dosages up to 150 mg/kg, proliferative changes in the Leydig cells of the testes, including benign neoplasm, were increased compared to control rats.
Other Effects: No systemic effects of lansoprazole on the central nervous system, lymphoid, hematopoietic, renal, hepatic, cardiovascular, or respiratory systems have been found in humans. Among 56 patients who had extensive baseline eye evaluations, no visual toxicity was observed after lansoprazole treatment (up to 180 mg/day) for up to 58 months. After lifetime lansoprazole exposure in rats, focal pancreatic atrophy, diffuse lymphoid hyperplasia in the thymus, and spontaneous retinal atrophy were seen.
Microbiology: Lansoprazole, clarithromycin and/or amoxicillin have been shown to be active against most strains of
Helicobacter pylori in vitro and in clinical infections as described in the Indications section (see Indications).
Helicobacter pylori Pretreatment Resistance: Clarithromycin pretreatment resistance (≥2.0 mcg/mL) was 9.5% (91/960) by E-test and 11.3% (12/106) by agar dilution in the dual and triple therapy clinical trials (M93-125, M93-130, M93-131, M95-392, and M95-399).
Amoxicillin pretreatment susceptible isolates (≤0.25 mcg/mL) occurred in 97.8% (936/957) and 98.0% (98/100) of the patients in the dual and triple therapy clinical trials by E-test and agar dilution, respectively. Twenty-one of 957 patients (2.2%) by E-test, and two of 100 patients (2.0%) by agar dilution, had amoxicillin pretreatment MICs of greater than 0.25 mcg/mL. One patient on the 14-day triple therapy regimen had an unconfirmed pretreatment amoxicillin minimum inhibitory concentration (MIC) of greater than 256 mcg/mL by E-test and the patient was eradicated of
H. pylori (see Table 3).
Click on icon to see table/diagram/image
Patients not eradicated of
H. pylori following lansoprazole/amoxicillin/clarithromycin triple therapy will likely have clarithromycin resistant H.pylori. Therefore, for those patients who fail therapy, clarithromycin susceptibility testing should be done when possible. Patients with clarithromycin resistant
H. pylori should not be treated with lansoprazole/amoxicillin/clarithromycin triple therapy or with regimens which include clarithromycin as the sole antimicrobial agent.
Amoxicillin Susceptibility Test Results and Clinical/Bacteriological Outcomes: In the dual and triple therapy clinical trials, 82.6% (195/236) of the patients that had pretreatment amoxicillin susceptible MICs (≤0.25 mcg/mL) were eradicated of H. pylori. Of those with pretreatment amoxicillin MICs of greater than 0.25 mcg/mL, three of six had the
H. pylori eradicated. A total of 30% (21/70) of the patients failed lansoprazole 30 mg three times daily/amoxicillin 1 g three times daily dual therapy and a total of 12.8% (22/172) of the patients failed the 10- and 14-day triple therapy regimens. Post-treatment susceptibility results were not obtained on 11 of the patients who failed therapy. Nine of the 11 patients with amoxicillin post-treatment MICs that failed the triple therapy regimen also had clarithromycin resistant H. pylori isolates.
Susceptibility Test for
Helicobacter pylori: For susceptibility testing information about
Helicobacter pylori, see Microbiology section in prescribing information for clarithromycin and amoxicillin.
Clinical Studies: Duodenal Ulcer: In a U.S. multicenter, double-blind, placebo-controlled, dose-response (15, 30, and 60 mg of TAKEPRON once daily) study of 284 patients with endoscopically documented duodenal ulcer, the percentage of patients healed after two and four weeks was significantly higher with all doses of TAKEPRON than with placebo. There was no evidence of a greater or earlier response with the two higher doses compared with TAKEPRON 15 mg.
Based on this study and the second study described as follows, the recommended dose of TAKEPRON in duodenal ulcer is 15 mg per day (see Table 4).
Click on icon to see table/diagram/image
TAKEPRON 15 mg was significantly more effective than placebo in relieving day and nighttime abdominal pain and in decreasing the amount of antacid taken per day.
In a second U.S. multicenter study, also double-blind, placebo-controlled, dose-comparison (15 and 30 mg of TAKEPRON once daily), and including a comparison with ranitidine, in 280 patients with endoscopically documented duodenal ulcer, the percentage of patients healed after four weeks was significantly higher with both doses of TAKEPRON than with placebo. There was no evidence of a greater or earlier response with the higher dose of TAKEPRON. Although the 15 mg dose of TAKEPRON was superior to ranitidine at four weeks, the lack of significant difference at two weeks and the absence of a difference between 30 mg of TAKEPRON and ranitidine leaves the comparative effectiveness of the two agents undetermined (see Table 5) (see Indications).
Click on icon to see table/diagram/image
H. pylori Eradication to Reduce the Risk of Duodenal Ulcer Recurrence: Randomized, double-blind clinical studies performed in the U.S. in patients with
H. pylori and duodenal ulcer disease (defined as an active ulcer or history of an ulcer within one year) evaluated the efficacy of TAKEPRON in combination with amoxicillin and clarithromycin as triple 14-day therapy or in combination with amoxicillin as dual 14-day therapy for the eradication of
H. pylori. Based on the results of these studies, the safety and efficacy of two different eradication regimens were established: Triple therapy: TAKEPRON 30 mg twice daily/amoxicillin 1 g twice daily/clarithromycin 500 mg twice daily; Dual therapy: TAKEPRON 30 mg three times daily/amoxicillin 1 g three times daily.
All treatments were for 14 days.
H. pylori eradication was defined as two negative tests (culture and histology) at four to six weeks following the end of treatment.
Triple therapy was shown to be more effective than all possible dual therapy combinations. Dual therapy was shown to be more effective than both monotherapies. Eradication of
H. pylori has been shown to reduce the risk of duodenal ulcer recurrence.
A randomized, double-blind clinical study performed in the U.S. in patients with
H. pylori and duodenal ulcer disease (defined as an active ulcer or history of an ulcer within one year) compared the efficacy of TAKEPRON triple therapy for 10 and 14 days. This study established that the 10-day triple therapy was equivalent to the 14-day triple therapy in eradicating
H. pylori (see Tables 6 and 7) (see Indications).
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Long-Term Maintenance Treatment of Duodenal Ulcers: TAKEPRON has been shown to prevent the recurrence of duodenal ulcers. Two independent, double-blind, multicenter, controlled trials were conducted in patients with endoscopically confirmed healed duodenal ulcers. Patients remained healed significantly longer and the number of recurrences of duodenal ulcers was significantly less in patients treated with TAKEPRON than in patients treated with placebo over a 12-month period (see Table 8) (see Indications).
Click on icon to see table/diagram/image
In trial #2, no significant difference was noted between TAKEPRON 15 mg and 30 mg in maintaining remission.
Gastric Ulcer: In a U.S. multicenter, double-blind, placebo-controlled study of 253 patients with endoscopically documented gastric ulcer, the percentage of patients healed at four and eight weeks was significantly higher with TAKEPRON 15 mg and 30 mg once a day than with placebo (see Table 9) (see Short-Term Treatment of Active Benign Gastric Ulcer under Indications).
Click on icon to see table/diagram/image
Patients treated with any TAKEPRON dose reported significantly less day and night abdominal pain along with fewer days of antacid use and fewer antacid tablets used per day than the placebo group.
Independent substantiation of the effectiveness of TAKEPRON 30 mg was provided by a meta-analysis of published and unpublished data.
Healing of NSAID-Associated Gastric Ulcer: In two U.S. and Canadian multicenter, double-blind, active-controlled studies in patients with endoscopically confirmed NSAID-associated gastric ulcer who continued their NSAID use, the percentage of patients healed after eight weeks was statistically significantly higher with 30 mg of TAKEPRON than with the active control. A total of 711 patients were enrolled in the study, and 701 patients were treated. Patients ranged in age from 18 to 88 years (median age 59 years), with 67% female patients and 33% male patients. Race was distributed as follows: 87% Caucasian, 8% Black, 5% Other. There was no statistically significant difference between TAKEPRON 30 mg daily and the active control on symptom relief (i.e., abdominal pain) (see Table 10) (see Healing of NSAID-Associated Gastric Ulcer under Indications).
Click on icon to see table/diagram/image
Risk Reduction of NSAID-Associated Gastric Ulcer: In one large U.S., multicenter, double-blind, placebo- and misoprostol-controlled (misoprostol blinded only to the endoscopist) study inpatients who required chronic use of an NSAID and who had a history of an endoscopically documented gastric ulcer, the proportion of patients remaining free from gastric ulcer at four, eight, and 12 weeks was significantly higher with 15 or 30 mg of TAKEPRON than placebo. A total of 537 patients were enrolled in the study, and 535 patients were treated. Patients ranged in age from 23 to 89 years (median age 60 years), with 65% female patients and 35% male patients. Race was distributed as follows: 90% Caucasian, 6% Black, 4% other. The 30 mg dose of TAKEPRON demonstrated no additional benefit in risk reduction of the NSAID-associated gastric ulcer than the 15 mg dose (see Table 11) (see Risk Reduction of NSAID-Associated Gastric Ulcer under Indications).
Click on icon to see table/diagram/image
Gastroesophageal Reflux Disease (GERD): Symptomatic GERD: In a U.S. multicenter, double-blind, placebo-controlled study of 214 patients with frequent GERD symptoms, but no esophageal erosions by endoscopy, significantly greater relief of heartburn associated with GERD was observed with the administration of lansoprazole 15 mg once daily up to eight weeks than with placebo. No significant additional benefit from lansoprazole 30 mg once daily was observed.
The intent-to-treat analyses demonstrated significant reduction in frequency and severity of day and night heartburn. Data for frequency and severity for the eight-week treatment period are presented in Table 12 and in Figures 1 and 2: (See Table 12 and Figures 1 and 2.)
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In two U.S., multicenter double-blind, ranitidine-controlled studies of 925 total patients with frequent GERD symptoms, but no esophageal erosions by endoscopy, lansoprazole 15 mg was superior to ranitidine 150 mg (twice daily) in decreasing the frequency and severity of day and night heartburn associated with GERD for the eight-week treatment period. No significant additional benefit from lansoprazole 30 mg once daily was observed (see Gastroesophageal Reflux Disease under Indications).
Erosive Esophagitis: In a U.S. multicenter, double-blind, placebo-controlled study of 269 patients entering with an endoscopic diagnosis of esophagitis with mucosal grading of two or more and grades three and four signifying erosive disease, the percentages of patients with healing are presented in Table 13: (See Table 13.)
Click on icon to see table/diagram/image
In this study, all TAKEPRON groups reported significantly greater relief of heartburn and less day and night abdominal pain along with fewer days of antacid use and fewer antacid tablets taken per day than the placebo group. Although all doses were effective, the earlier healing in the higher two doses suggests 30 mg daily as the recommended dose.
TAKEPRON was also compared in a U.S. multicenter, double-blind study to a low dose of ranitidine in 242 patients with erosive reflux esophagitis. TAKEPRON at a dose of 30 mg was significantly more effective than ranitidine 150 mg twice daily as shown as follows (see Table 14).
Click on icon to see table/diagram/image
In addition, patients treated with TAKEPRON reported less day and nighttime heartburn and took less antacid tablets for fewer days than patients taking ranitidine 150 mg twice daily.
Although this study demonstrates effectiveness of TAKEPRON in healing erosive esophagitis, it does not represent an adequate comparison with ranitidine because the recommended ranitidine dose for esophagitis is 150 mg four times daily, twice the dose used in this study.
In the two trials described and in several smaller studies involving patients with moderate to severe erosive esophagitis, TAKEPRON produced healing rates similar to those shown previously.
In a U.S. multicenter, double-blind, active-controlled study, 30 mg of TAKEPRON was compared with ranitidine 150 mg twice daily in 151 patients with erosive reflux esophagitis that was poorly responsive to a minimum of 12 weeks of treatment with at least one H
2-receptor antagonist given at the dose indicated for symptom relief or greater, namely, cimetidine 800 mg/day, ranitidine 300 mg/day, famotidine 40 mg/day or nizatidine 300 mg/day.
TAKEPRON 30 mg was more effective than ranitidine 150 mg twice daily in healing reflux esophagitis, and the percentage of patients with healing were as follows. This study does not constitute a comparison of the effectiveness of histamine H
2-receptor antagonists with TAKEPRON, as all patients had demonstrated unresponsiveness to the histamine H
2-receptor antagonist mode of treatment. It does indicate, however, that TAKEPRON may be useful in patients failing on a histamine H
2-receptor antagonist (see Table 15) (see Gastroesophageal Reflux Disease (GERD) under Indications).
Click on icon to see table/diagram/image
Long-Term Maintenance Treatment of Erosive Esophagitis: Two independent, double-blind, multicenter, controlled trials were conducted in patients with endoscopically confirmed healed esophagitis. Patients remained in remission significantly longer and the number of recurrences of erosive esophagitis was significantly less in patients treated with TAKEPRON than in patients treated with placebo over a 12-month period (see Table 16).
Click on icon to see table/diagram/image
Regardless of initial grade of erosive esophagitis, TAKEPRON 15 mg and 30 mg were similar in maintaining remission.
In a U.S., randomized, double-blind, study, TAKEPRON 15 mg daily (n = 100) was compared with ranitidine 150 mg twice daily (n = 106),at the recommended dosage, in patients with endoscopically-proven healed erosive esophagitis over a 12-month period. Treatment with TAKEPRON resulted in patients remaining healed (Grade 0 lesions) of erosive esophagitis for significantly longer periods of time than those treated with ranitidine (p<0.001). In addition, TAKEPRON was significantly more effective than ranitidine in providing complete relief ofboth daytime and nighttime heartburn. Patients treated with TAKEPRON remained asymptomatic for a significantly longer period of time than patients treated with ranitidine (see Maintenance of Healing of Erosive Esophagitis under Indications).
Pathological Hypersecretory Conditions Including Zollinger-Ellison Syndrome: In open studies of 57 patients with pathological hypersecretory conditions, such as Zollinger-Ellison syndrome (ZES) with or without multiple endocrine adenomas, TAKEPRON significantly inhibited gastric acid secretion and controlled associated symptoms of diarrhea, anorexia and pain. Doses ranging from 15 mg every other day to 180 mg per day maintained basal acid secretion below 10 mEq/hr in patients without prior gastric surgery and below 5 mEq/hr in patients with prior gastric surgery.
Initial doses were titrated to the individual patient need, and adjustments were necessary with time in some patients (see Dosage & Administration). TAKEPRON was well tolerated at these high dose levels for prolonged periods (greater than four years in some patients). In most ZES patients, serum gastrin levels were not modified by TAKEPRON. However, in some patients, serum gastrin increased to levels greater than those present prior to initiation of lansoprazole therapy (see Pathological Hypersecretory Conditions Including Zollinger-Ellison Syndrome (ZES) under Indications).
Pharmacokinetics: TAKEPRON OD Tablets contain an enteric-coated granule formulation of lansoprazole. Absorption of lansoprazole begins only after the granules leave the stomach. Absorption is rapid, with mean peak plasma levels of lansoprazole occurring after approximately 1.7 hours. After a single-dose administration of 15 mg to 60 mg of oral lansoprazole, the peak plasma concentrations (C
max) of lansoprazole and the area under the plasma concentration curves (AUCs) of lansoprazole were approximately proportional to theadministered dose. Lansoprazole does not accumulate and its pharmacokinetics are unaltered by multiple dosing.
Absorption: The absorption of lansoprazole is rapid, with the mean C
max occurring approximately 1.7 hours after oral dosing, and the absolute bioavailability is over 80%. In healthy subjects, the mean (±SD) plasma half-life was 1.5 (±1.0) hours. Both the C
max and AUC are diminished by about 50% to 70% if lansoprazole is given 30 minutes after food, compared to the fasting condition. There is no significant food effect if lansoprazole is given before meals.
Distribution: Lansoprazole is 97% bound to plasma proteins. Plasma protein binding is constant over the concentration range of 0.05 to 5.0 mcg/mL.
Metabolism: Lansoprazole is extensively metabolized in the liver. Two metabolites have been identified in measurable quantities in plasma (the hydroxylated sulfinyl and sulfone derivatives of lansoprazole). These metabolites have very little or no antisecretory activity. Lansoprazole is thought to be transformed into two active species which inhibit acid secretion by blocking the proton pump [(H
+, K
+)-ATPase enzyme system] at the secretory surface of the gastric parietal cell. The two active species are not present in the systemic circulation. The plasma elimination half-life of lansoprazole is less than two hours while the acid inhibitory effect lasts more than 24 hours. Therefore, the plasma elimination half-life of lansoprazole does not reflect its duration of suppression of gastric acid secretion.
Elimination: Following single-dose oral administration of TAKEPRON, virtually no unchanged lansoprazole was excreted in the urine. In one study, after a single oral dose of
14C-lansoprazole, approximately one-third of the administered radiation was excreted in the urine and two-thirds was recovered in the feces. This implies a significant biliary excretion of the lansoprazole metabolites.
Specific Populations: Pediatric Use: One to 17 years of age: The pharmacokinetics of lansoprazole were studied in pediatric patients with GERD aged one to 11 years and 12 to 17 years in two separate clinical studies. In children aged one to 11 years, lansoprazole was dosed 15 mg daily for subjects weighing ≤30 kg and 30 mg daily for subjects weighing greater than 30 kg. Mean C
max and AUC values observed on Day 5 of dosing were similar between the two dose groups and were not affected by weight or age within each weight-adjusted dose group used in the study. In adolescent subjects aged 12 to 17 years, subjects were randomized to receive lansoprazole at 15 mg or 30 mg daily. Mean C
max and AUC values of lansoprazole were not affected by body weight or age; and nearly dose-proportional increases in mean C
max and AUC values were observed between the two dose groups in the study. Overall, lansoprazole pharmacokinetics in pediatric patients aged 1 to 17 years were similar to those observed in healthy adult subjects.
Neonate to less than one year of age: Refer to Use in Children under Precautions for the pharmacokinetics of lansoprazole in pediatric patients with GERD aged less than 28 days and one to 11 months.
Geriatric Use: The clearance of lansoprazole is decreased in the elderly, with elimination half-life increased approximately 50% to 100%. Because the mean half-life in the elderly remains between 1.9 to 2.9 hours, repeated once daily dosing does not result in accumulation of lansoprazole. Peak plasma levels were not increased in the elderly. No dosage adjustment is necessary in the elderly (see Precautions).
Renal Impairment: In patients with severe renal impairment, plasma protein binding decreased by 1.0% to 1.5% after administration of 60 mg of lansoprazole. Patients with renal impairment had a shortened elimination half-life and decreased total AUC (free and bound). The AUC for free lansoprazole in plasma, however, was not related to the degree of renal impairment; and the C
max and T
max (time to reach the maximum concentration) were not different than the C
max and T
max from subjects with normal renal function. No dosage adjustment is necessary inpatients with renal impairment (see Renal impairment under Precautions).
Hepatic Impairment: In patients with various degrees of chronic hepatic impairment, the mean plasma half-life of lansoprazole was prolonged from 1.5 hours to 3.2 to 7.2 hours. An increase in the mean AUC of up to 500% was observed at steady state in hepatically-impaired patients compared to healthy subjects. Consider dose reduction in patients with severe hepatic impairment (see Hepatic impairment under Precautions).
Gender: In a study comparing 12 male and six female human subjects who received lansoprazole, no gender differences were found in pharmacokinetics and intragastric pH results (see Gender under Precautions).
Drug-Drug Interactions: TAKEPRON may interfere with the absorption of other drugs where gastric pH is an important determinant of bioavailability (e.g., ketoconazole, ampicillin esters, iron salts, digoxin).
Lansoprazole is metabolized through the cytochrome P
450 system, specifically through the CYP3A and CYP2C19 isozymes. Studies have shown that TAKEPRON does not have clinically significant interactions with other drugs metabolized by the cytochrome P
450 system, such as warfarin, antipyrine, indomethacin, ibuprofen, phenytoin, propranolol, prednisone, diazepam, or clarithromycin in healthy subjects. These compounds are metabolized through various cytochrome P
450 isozymes including CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A.
Atazanavir and Nelfinavir: Lansoprazole causes long-lasting inhibition of gastric acid secretion. Lansoprazole substantially decreases the systemic concentrations of HIV protease inhibitors, such as atazanavir and nelfinavir, which are dependent upon the presence of gastric acid for absorption, and may result in a loss of therapeutic effect of atazanavir or nelfinavir and the development of HIV resistance. Therefore, TAKEPRON, or other proton pump inhibitors, should not be co-administered with atazanavir or nelfinavir.
Theophylline: When TAKEPRON was administered concomitantly with theophylline (CYP1A2, CYP3A), a minor increase (10%) in the clearance of theophylline was seen. Because of the small magnitude and the direction of the effect on theophylline clearance, this interaction is unlikely to be of clinical concern. Nonetheless, individual patients may require additional titration of their theophylline dosage when TAKEPRON is started or stopped to ensure clinically effective blood levels.
Warfarin: In a study of healthy subjects neither the pharmacokinetics of warfarin enantiomers nor prothrombin time were affected following single or multiple 60 mg doses of lansoprazole. However, there have been reports of increased International Normalized Ratio (INR) and prothrombin time in patients receiving proton pump inhibitors, including TAKEPRON, and warfarin concomitantly. Increases in INR and prothrombin time may lead to abnormal bleeding and even death. Patients treated with proton pump inhibitors and warfarin concomitantly may need to be monitored for increases in INR and prothrombin time.
Methotrexate and 7-hydromethotrexate: In an open-label, single-arm, eight-day, pharmacokinetic study of 28 adult rheumatoid arthritis patients (who required the chronic use of 7.5 to 15 mg of methotrexate given weekly), administration of seven days of naproxen 500 mg twice daily and TAKEPRON 30 mg daily had no effect on the pharmacokinetics of methotrexate and 7-hydroxymethotrexate. While this study was not designed to assess the safety of this combination of drugs, no major adverse reactions were noted. However, this study was conducted with low doses of methotrexate. A drug interaction study with high doses of methotrexate has not been conducted.
Amoxicillin: TAKEPRON has also been shown to have no clinically significant interaction with amoxicillin.
Sucralfate: In a single-dose crossover study examining TAKEPRON 30 mg and omeprazole 20 mg each administered alone and concomitantly with sucralfate 1 gram, absorption of the proton pump inhibitors was delayed and their bioavailability was reduced by 17% and 16%, respectively, when administered concomitantly with sucralfate. Therefore, proton pump inhibitors should be taken at least 30 minutes prior to sucralfate. In clinical trials, antacids were administered concomitantly with TAKEPRON and there was no evidence of a change in the efficacy of TAKEPRON.
Clopidogrel: Clopidogrel is metabolized to its active metabolite in part by CYP2C19. A study of healthy subjects who were CYP2C19 extensive metabolizers, receiving once daily administration of clopidogrel 75 mg alone or concomitantly with TAKEPRON 30 mg (n=40),for nine days was conducted. The mean AUC of the active metabolite of clopidogrel was reduced by approximately 14% (mean AUC ratio was 86%, with 90% CI of 80 to 92%) when TAKEPRON was coadministered compared to administration of clopidogrel alone. Pharmacodynamic parameters were also measured and demonstrated that the change in inhibition of platelet aggregation (induced by 5 mcM ADP) was related to the change in the exposure to clopidogrel active metabolite. The clinical significance of this finding is not clear.
Nonclinical Toxicology: Carcinogenesis, Mutagenesis, Impairment of Fertility: In two 24-month carcinogenicity studies, Sprague-Dawley rats were treated with oral lansoprazole doses of five to 150 mg/kg/day, about one to 40 times the exposure on a body surface (mg/m
2) basis of a 50 kg person of average height [1.46 m
2 body surface area (BSA)] given the recommended human dose of 30 mg/day. Lansoprazole produced dose-related gastric enterochromaffin-like (ECL) cell hyperplasia and ECL cell carcinoids in both male and female rats. It also increased the incidence of intestinal metaplasia of the gastric epithelium in both sexes. In male rats, lansoprazole produced a dose-related increase of testicular interstitial cell adenomas. The incidence of these adenomas in rats receiving doses of 15 to 150 mg/kg/day (four to 40 times the recommended human dose based on BSA) exceeded the low background incidence (range = 1.4 to 10%) for this strain of rat.
In a 24-month carcinogenicity study, CD-1 mice were treated with oral lansoprazole doses of 15 to 600 mg/kg/day, two to 80 times there commended human dose based on BSA. Lansoprazole produced a dose-related increased incidence of gastric ECL cell hyperplasia. It also produced an increased incidence of liver tumors (hepatocellular adenoma plus carcinoma). The tumor incidences in male mice treated with 300 and 600 mg/kg/day (40 to 80 times the recommended human dose based on BSA) and female mice treated with 150 to 600 mg/kg/day (20 to 80 times the recommended human dose based on BSA) exceeded the ranges of background incidences in historical controls for this strain of mice. Lansoprazole treatment produced adenoma of rete testis in male mice receiving 75 to 600 mg/kg/day (10 to 80 times the recommended human dose based on BSA).
A 26-week p53 (+/-) transgenic mouse carcinogenicity study was not positive.
Lansoprazole was positive in the Ames test and the
in vitro human lymphocyte chromosomal aberration assay. Lansoprazole was not genotoxicin the
ex vivo rat hepatocyte unscheduled DNA synthesis (UDS) test, the
in vivo mouse micronucleus test, or the rat bone marrow cell chromosomal aberration test.
Lansoprazole at oral doses up to 150 mg/kg/day (40 times the recommended human dose based on BSA) was found to have no effect onfertility and reproductive performance of male and female rats.
Animal Toxicology and/or Pharmacology: Reproductive Toxicology Studies: Reproduction studies have been performed in pregnant rats at oral lansoprazole doses up to 150 mg/kg/day [40 times the recommended human dose (30 mg/day) based on body surface area (BSA)] and pregnant rabbits at oral lansoprazole doses up to 30 mg/kg/day (16 times the recommended human dose based on BSA) and have revealed no evidence of impaired fertility or harm to the fetus due to lansoprazole.


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image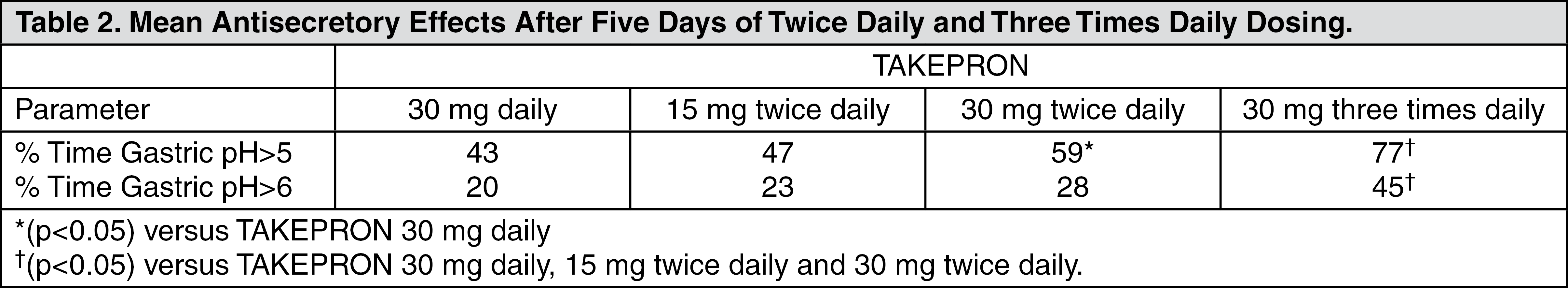 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image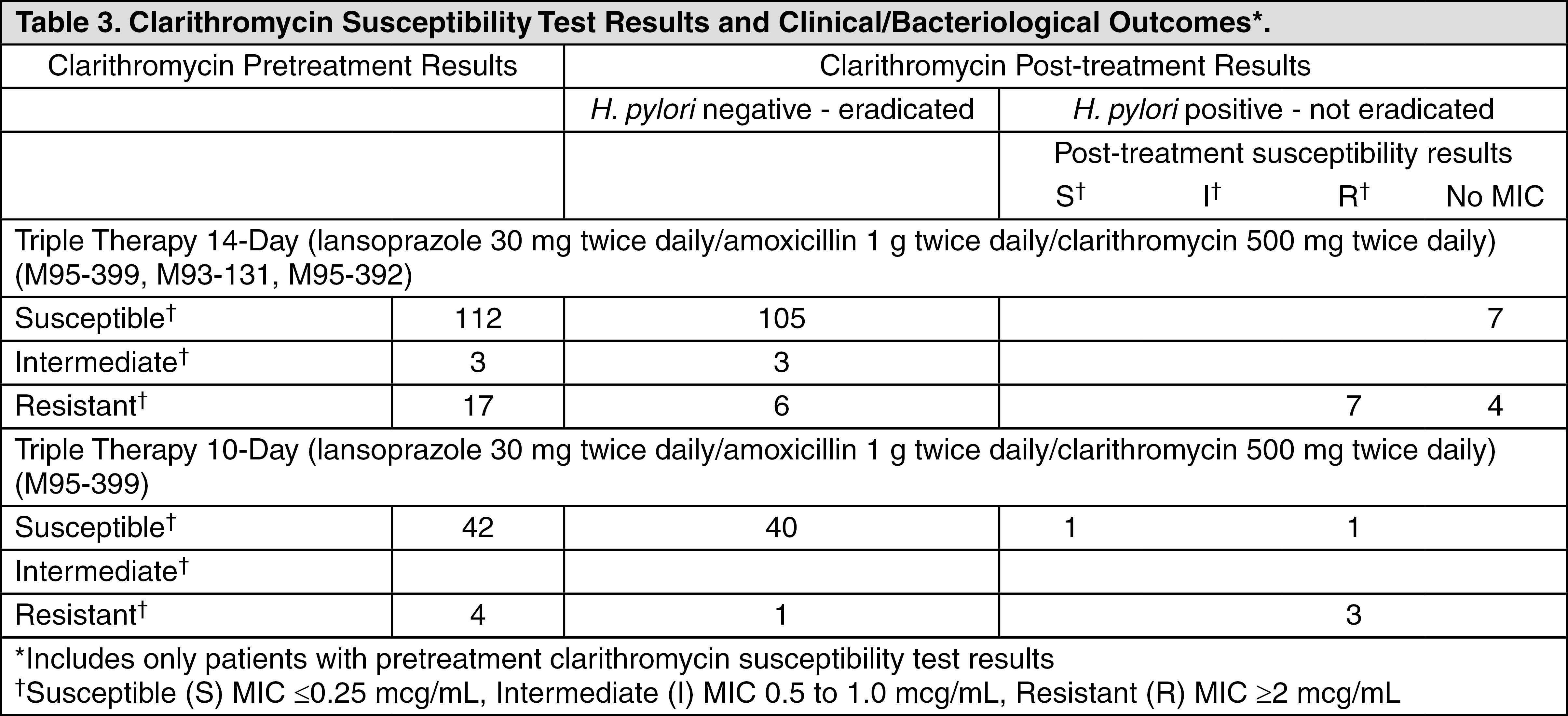 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image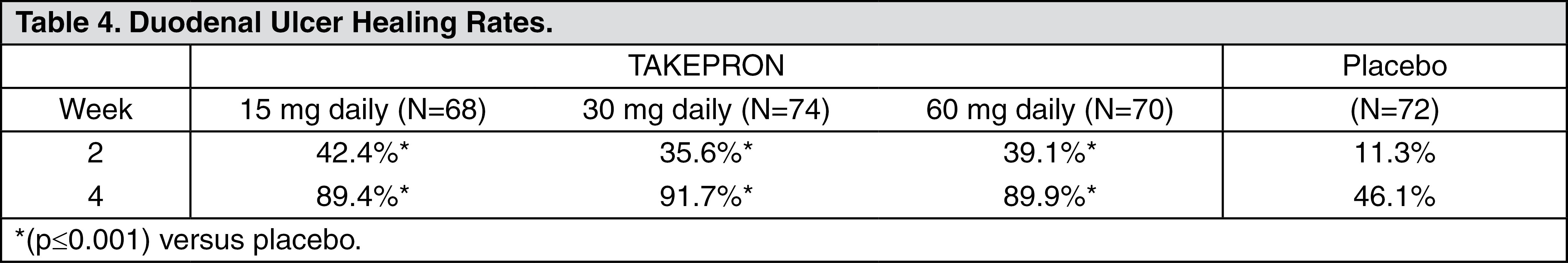 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image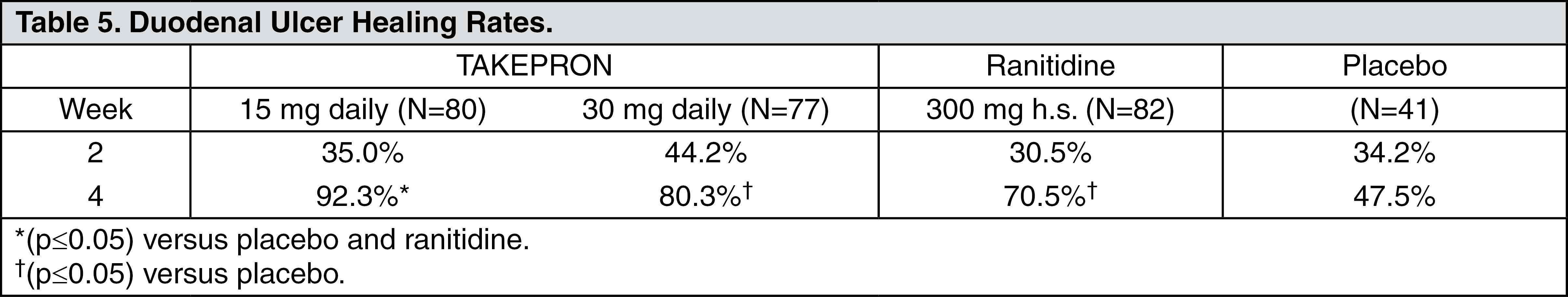 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image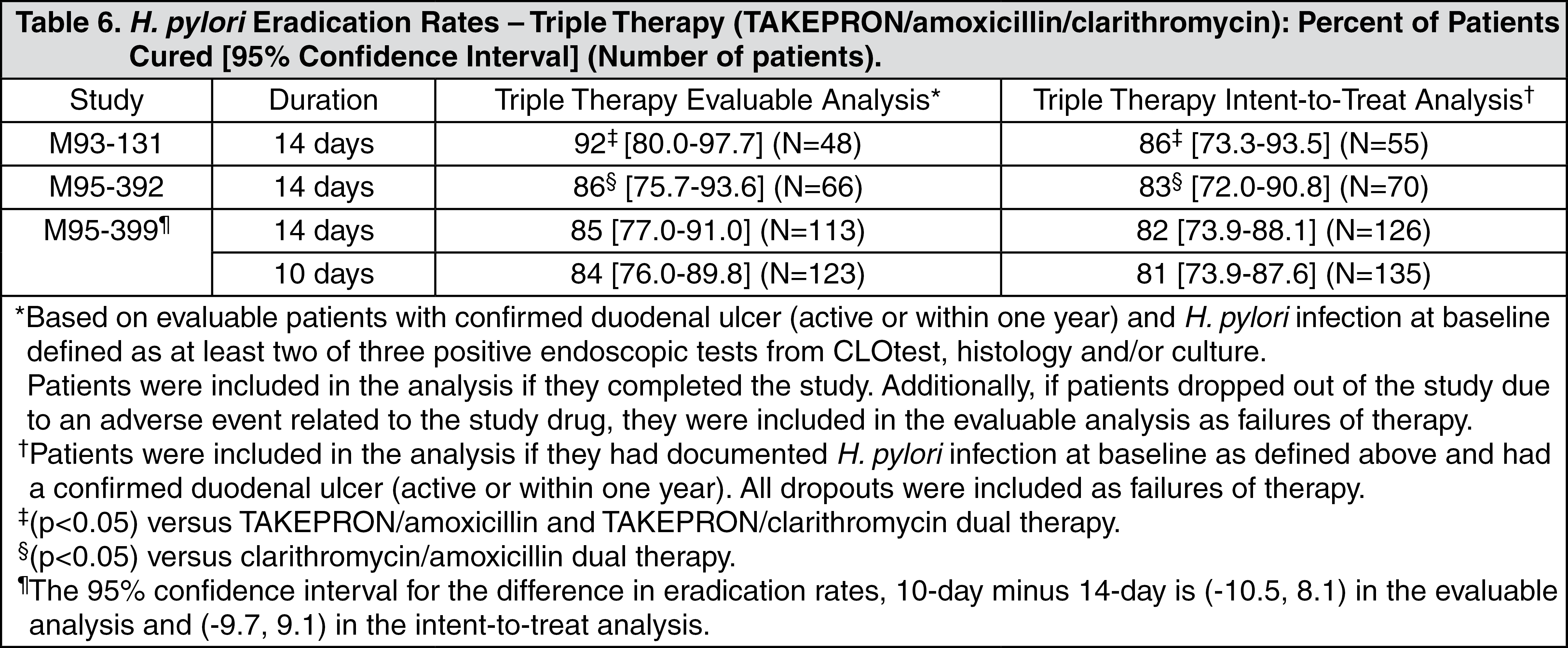 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image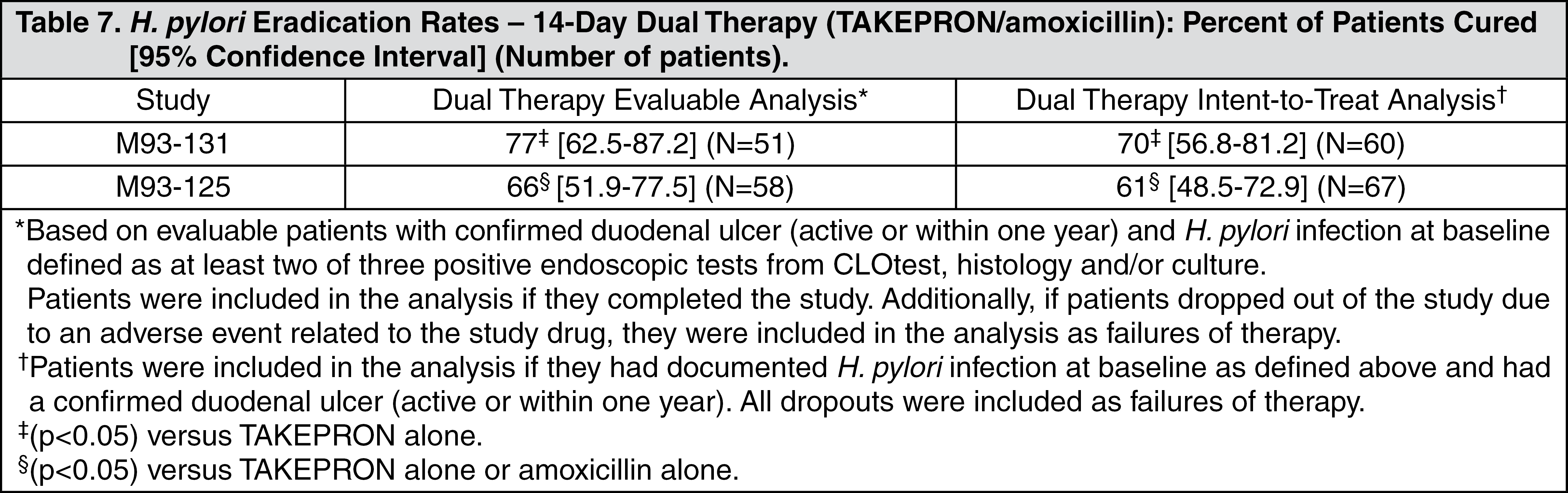 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image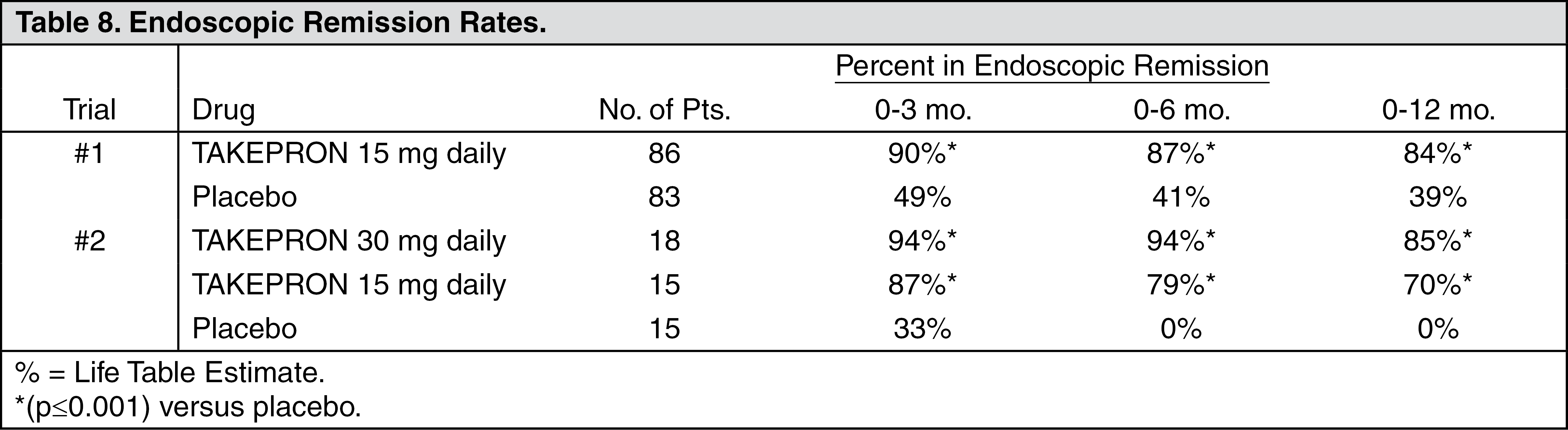 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image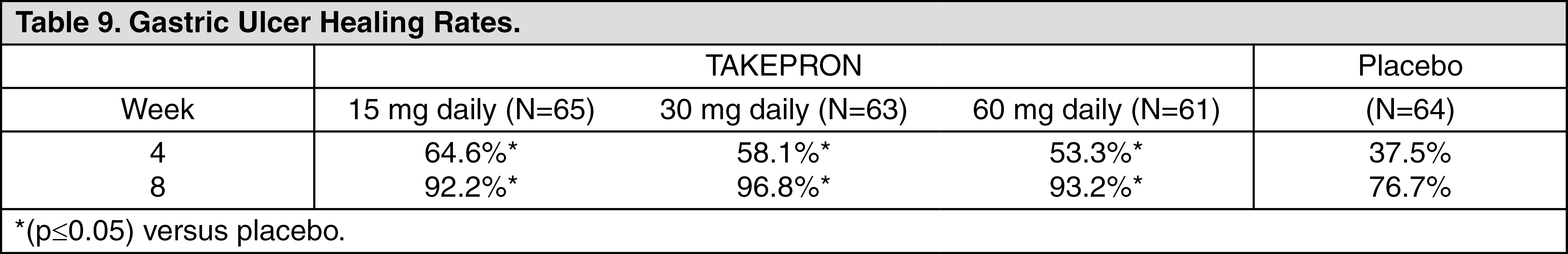 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image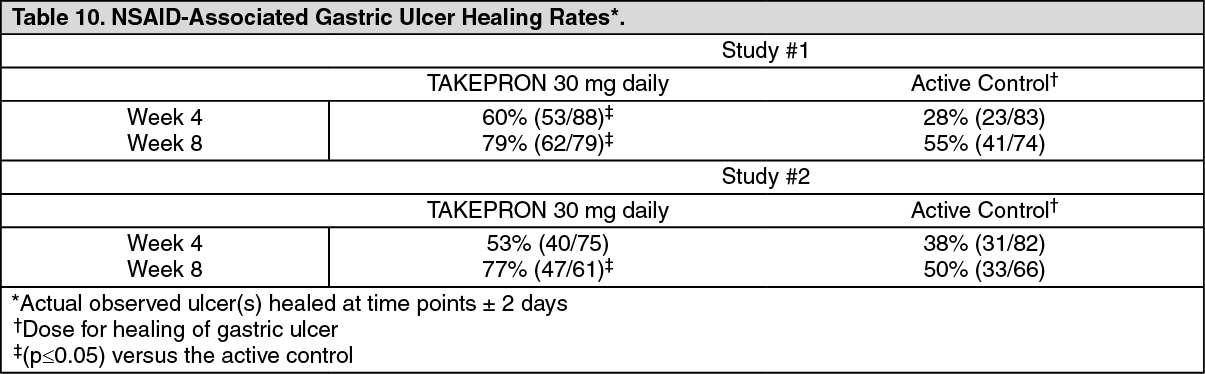 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image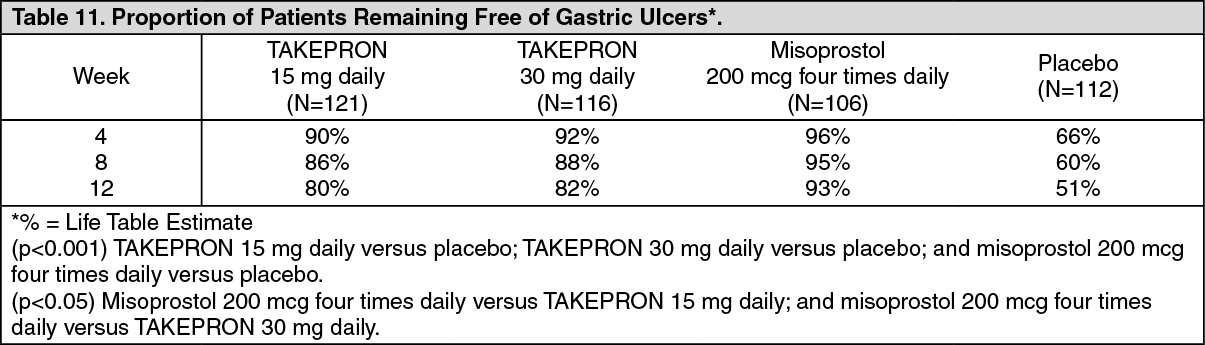 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image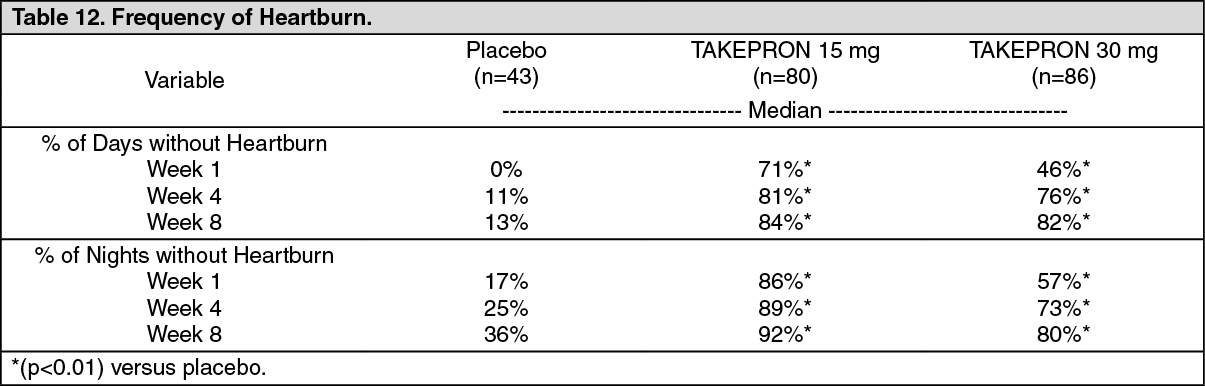 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image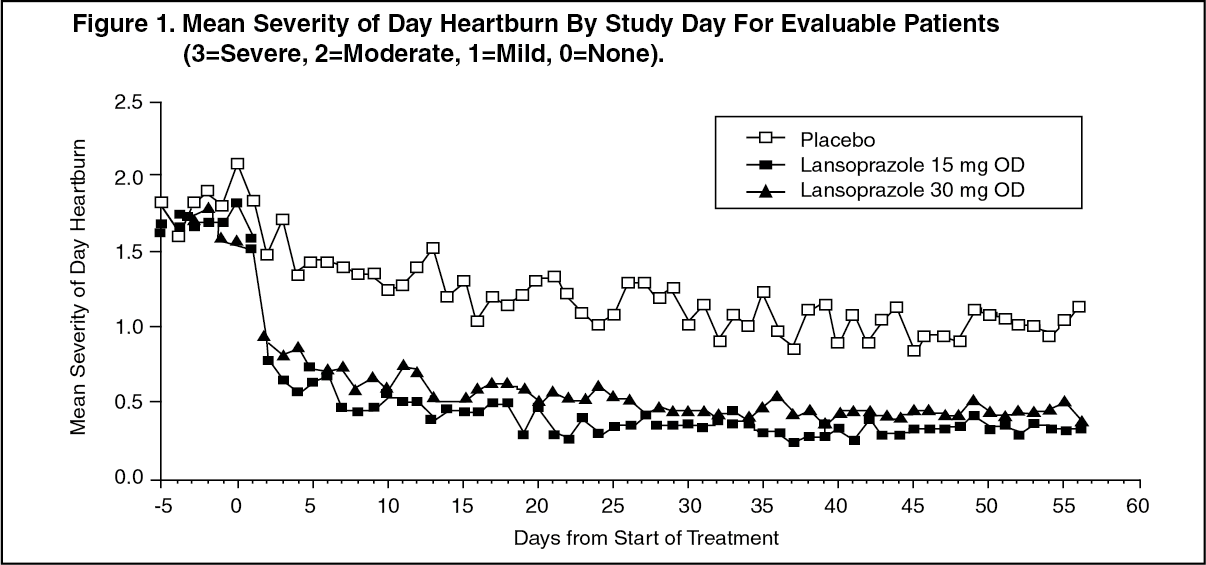 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image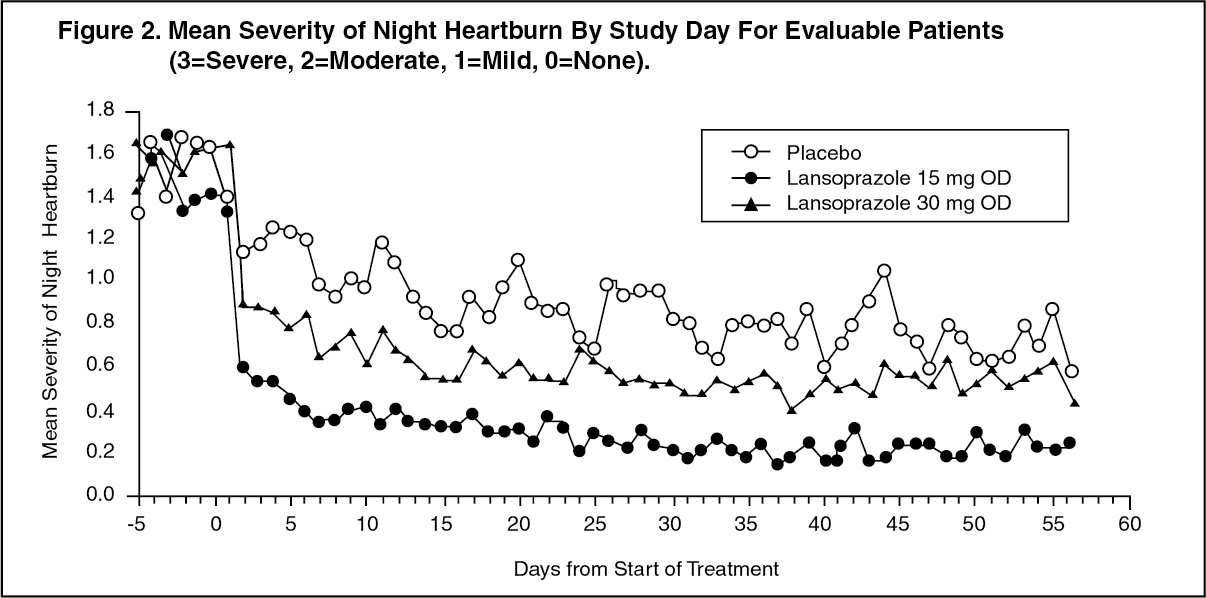 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image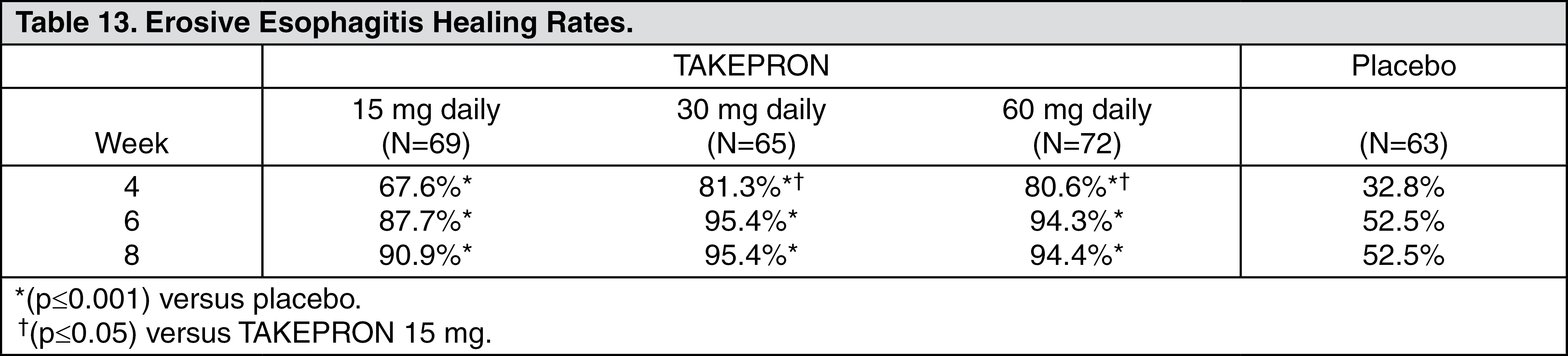 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image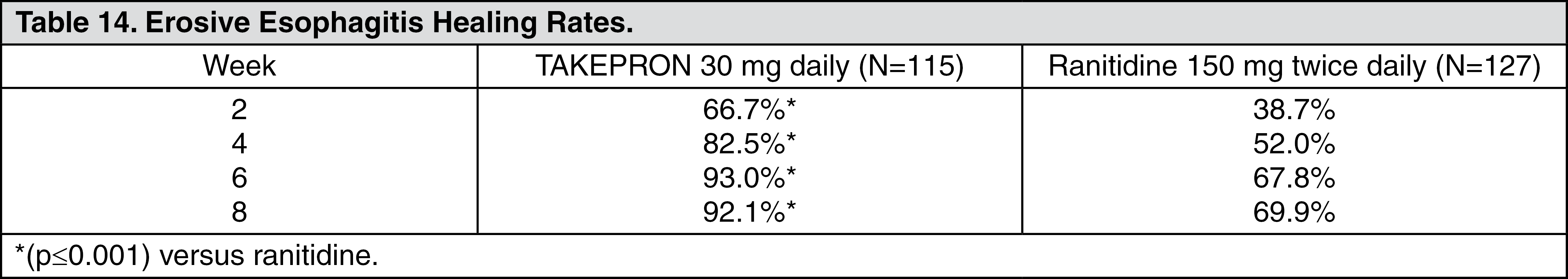 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image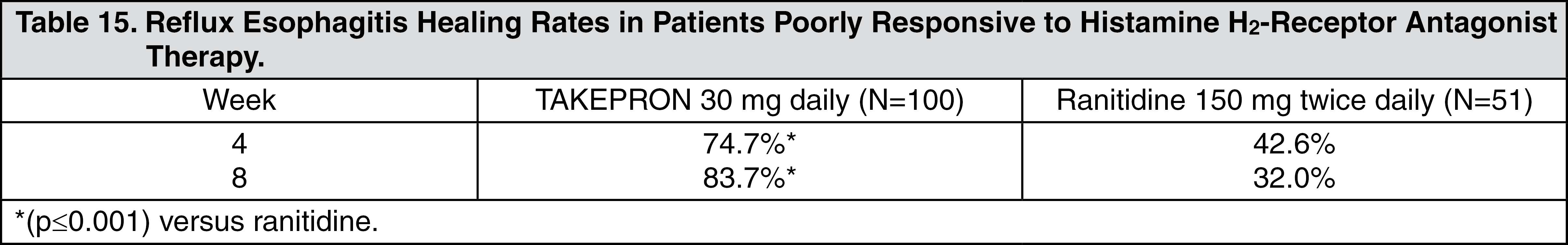 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image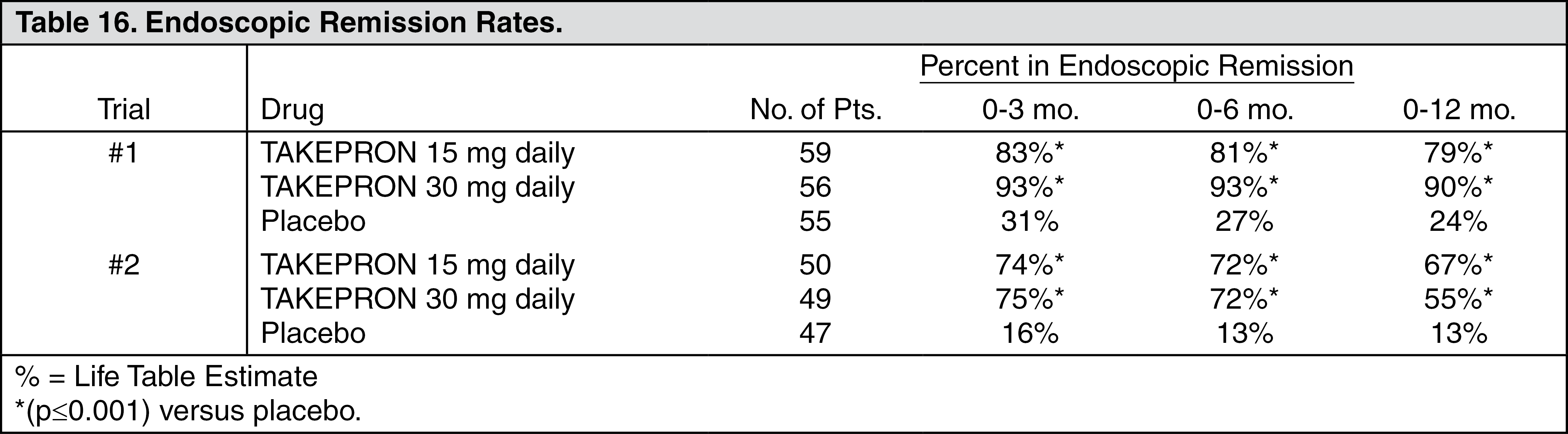 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image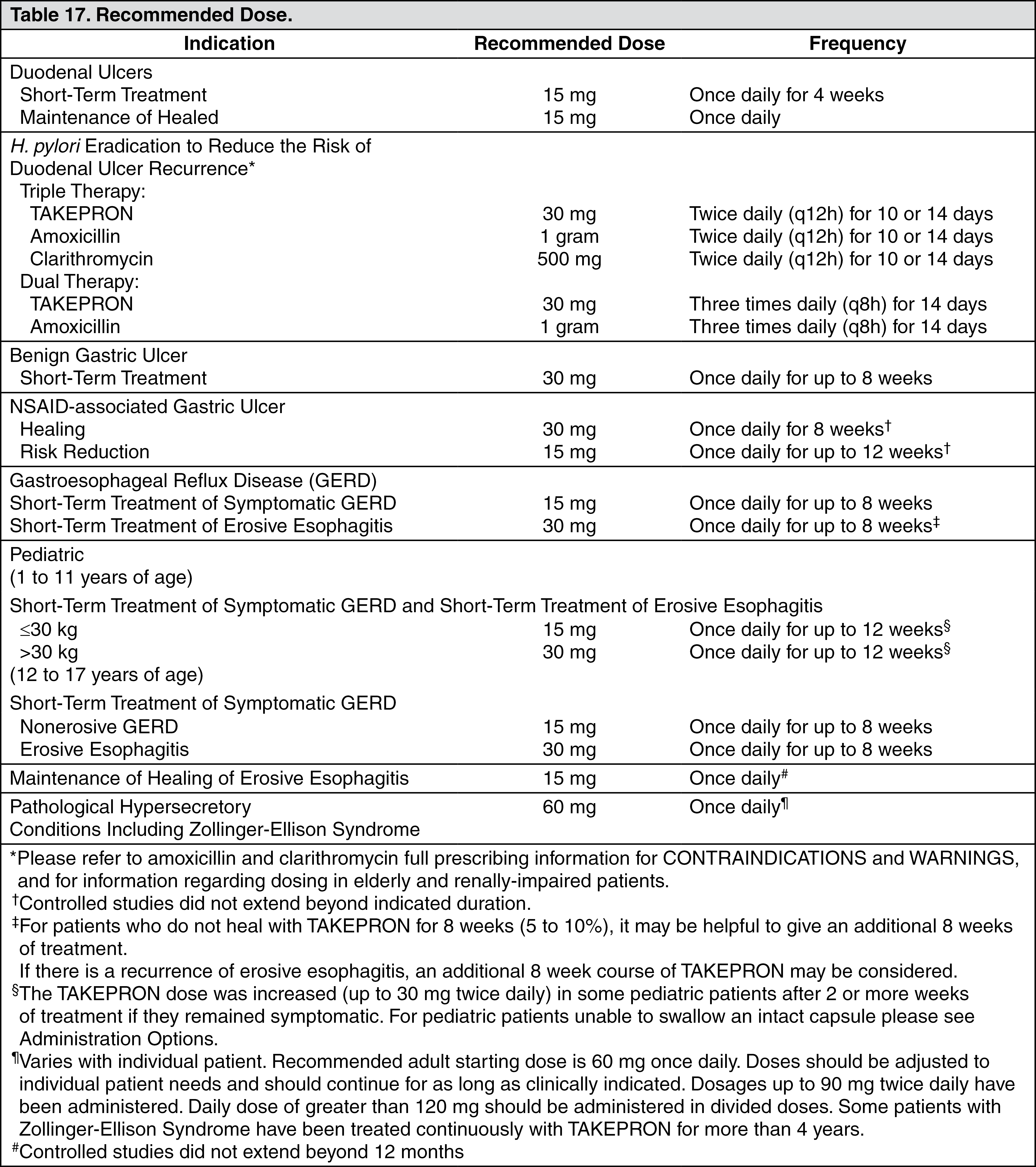 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image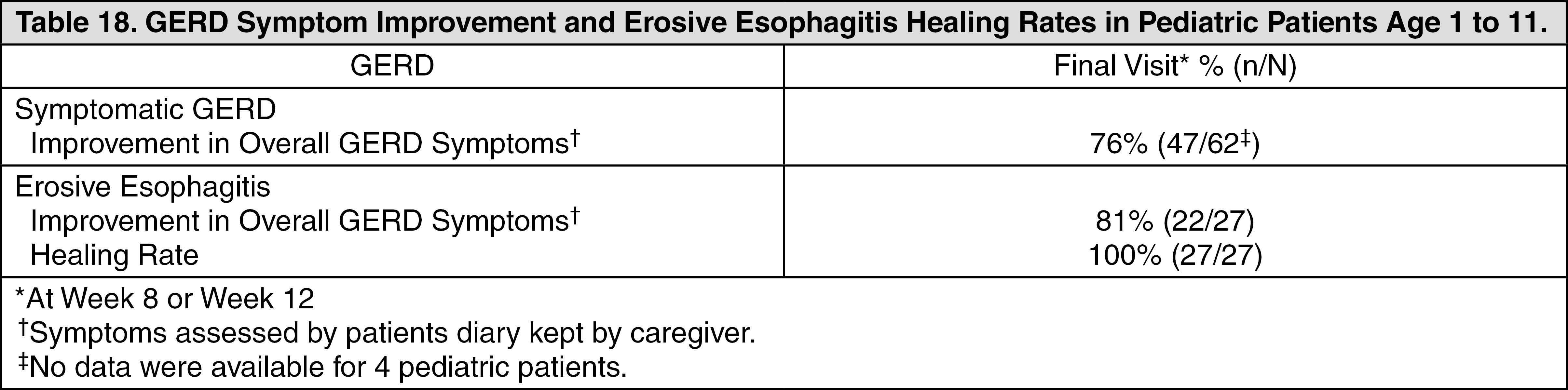 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image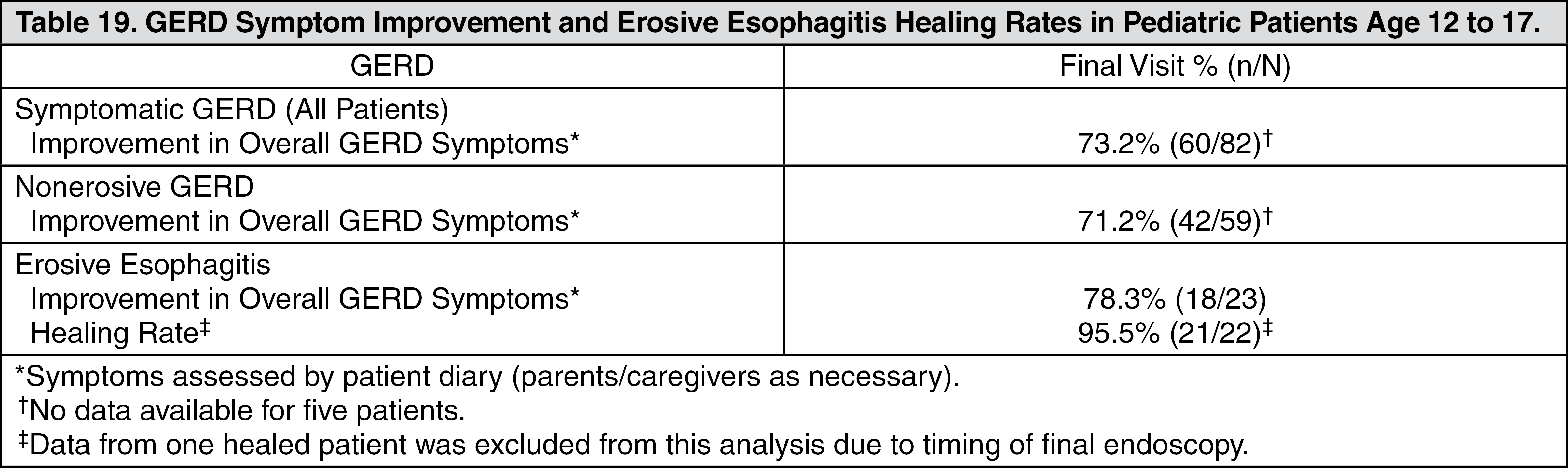 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image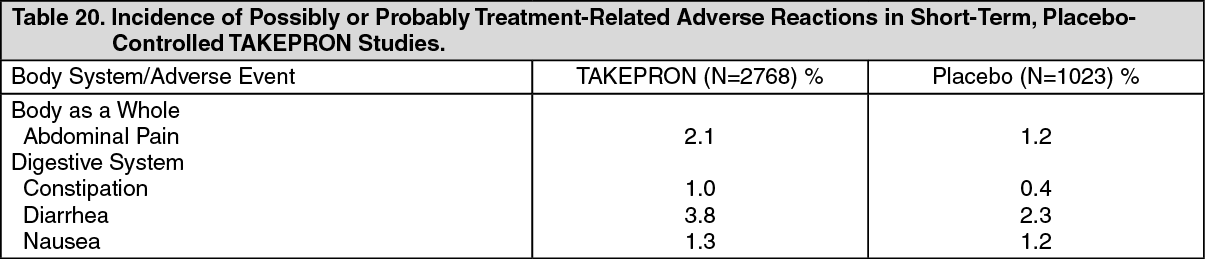 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image


 Sign Out
Sign Out